20240712更新:
软件作者看到了这篇教程, 指出其中错误, 并对分析挂载流程提出了相当多的宝贵意见, 具体内容已整合到一篇新的文章, 请移步查看.
以下原文:
HapHiC 是一款 reference free 的基因组 scaffolding 软件,论文预印本发表在 bioRxiv 上。论文一作 Xiaofei Zeng 在个人博客中做了一些 HapHiC 软件的系列教程以及使用技巧,虽然今年三月的时候还回复过评论,但是文章已经很久没更新了;
近期在尝试测试这个软件的时候发现找不到中文教程,不如自己写一篇,帮助有相同需求的朋友;
软件安装
推荐使用 conda 安装,不介绍手动安装的方式, 首先 clone 仓库到本地,我一般习惯 clone 到 conda/envs 目录下:
$ git clone https://github.com/zengxiaofei/HapHiC.git使用 clone 下来仓库中的 yml 文件构建 conda 环境:
conda env create -f HapHiC/conda_env/environment_py310.yml安装完成后,可以 soure 环境,也可以将构建环境的 bin 目录 export 到环境变量中,要是用的频繁就写到 .bashrc 里:
source conda/bin/activate haphic
# or
export PATH=/path/to/haphic/bin:$PATH软件自带检测工具,check 一下;注意,这个 haphic 主流程文件在 clone 下来的仓库目录下,不在 conda create 的环境下,我第一次就没找到:
/path/to/HapHiC/haphic checkcheck 通过,空运行软件弹出欢迎界面,使用 --help 查看参数即可;
/path/to/HapHiC/haphic --help
有几个点需要注意:
git clone下来的仓库名称是 HapHiC, 而基于 yml 文件构建的环境名称是 haphic; 如果你像我一样将仓库 clone 到了 conda/envs 目录下,这样这个目录会有 HapHiC 和 haphic 两个文件夹;如果介意请手动修改 yml 文件;- 部分 python 包是基于 pip 安装下载的,如果没有事先配置过 pip config, 下载会比较慢,部分包下载超时导致安装包错;建议事先配置 pip 的清华源;不同的 pip 用的是同一个 home 路径下的 config, 所以可以用一个已装好的 pip 配置一下;
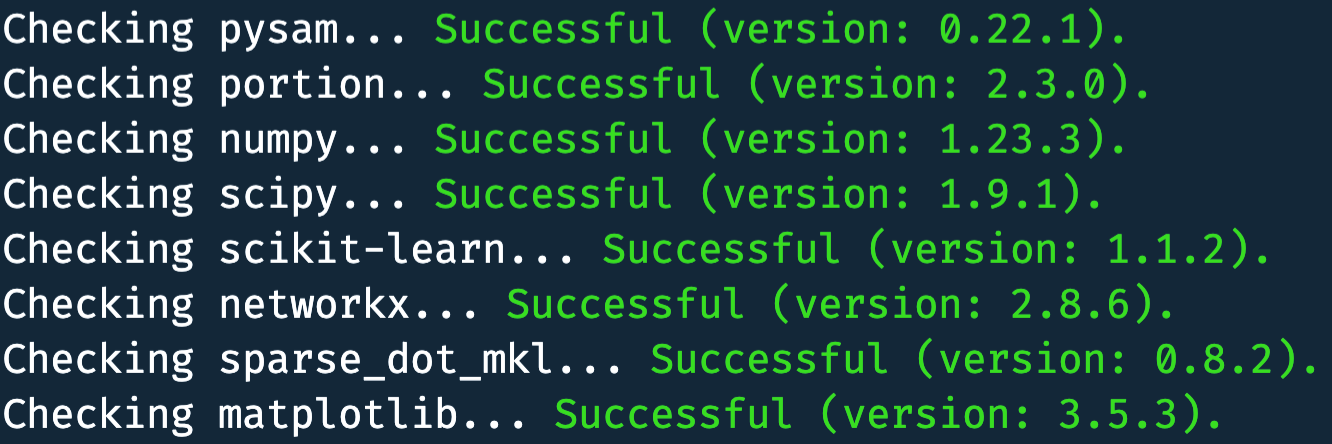
- 安装流程中第三步的 check 非常有必要,流程自动检查软件运行的关键包是否正确安装,哪个包失败
pip install一下就可以了;
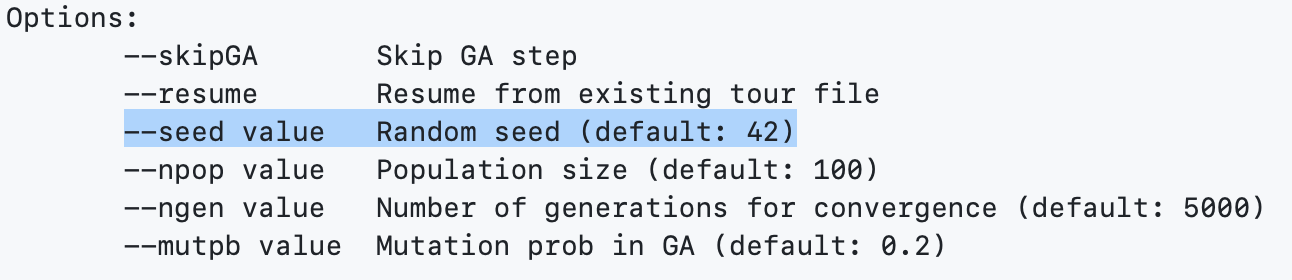
- 软件接口极多,部分接口和 ALLHiC 是一样的,如果之前用过 ALLHiC 可能容易理解一些;我之前没用过 ALLHiC, 啃这些接口有点吃力;发现 ALLHiC 作者还挺浪漫,用宇宙真理做随机种子:
- 软件默认基于 python 10, 所以对 gcc 版本以及 glib 库版本有一定要求,较老的集群应该无法安装;
软件使用
reads 比对
比对这里还是需要说一下:
基于 bwa 的比对
使用软件之前,需要事先准备比对好的结果文件,作者推荐使用 bwa 的比对方法:
$ bwa index asm.fa
$ bwa mem -5SP asm.fa /path/to/read1_fq.gz /path/to/read2_fq.gz | samblaster | samtools view - -@ 14 -S -h -b -F 3340 -o HiC.bam
$ /path/to/HapHiC/utils/filter_bam HiC.bam 1 --nm 3 --threads 14 | samtools view - -b -@ 14 -o HiC.filtered.bam我们拆解一下作者这里的意图:
-5SP: 在质量值低于 5 的碱基末端增加剪辑标记 S;samblaster: 一般用于去除 PCR dup;-F 3340: 过滤 PCR dup 和次比对;- 后面还使用自己封装的
filter_bam做进一步过滤,根据说明默认处理的是MAPQ ≥ 1和NM < 3情况;
基于 chromap 的比对
在 V 1.0.3 版本更新中,软件支持了 chromap 输出的 pairs 格式:
- Version 1.0.3 (2024.03.21): Add support for the pairs format used in chromap.
我个人非常喜欢 chromap 这个软件,单独说一下这里:
- chromap 比对本身支持
--remove-pcr-duplicates去除 PCR dup; - 生成 pairs 的比对速度远高于生成 sam 的速度(约 1/3), 这还不算后续处理 sam 的
view和sort的时间消耗;从时间成本上非常理想;
其他注意事项
这里有一点其他注意事项和建议:
- 毕竟是新软件,要考虑兼容已有分析流程;目前 HapHiC 仅支持 bam 文件格式及 pairs 文件格式作为比对结果输入,不支持如 bed 等其他比对结果格式;建议根据自己已有的流程决定具体使用 bam 还是 pairs;
- 对于 bam 文件,作者强调了** DO NOT sort it by coordinate**, 如果已有了之前处理过的 bam, 建议
sort -n一下避免后续流程报错;
HapHiC 分析
One-line command
软件分析本身分为四步,但支持 one-line command 一行命令出结果
- Clustering
- Reassignment
- Ordering and orientation
- Building pseudomolecules
如果有需要可以分步具体处理,这样还挺好的,针对性很强,异常中断也不需要从头再来了,这里仅介绍直出结果的模式:
$ /path/to/HapHiC/haphic pipeline asm.fa HiC.filtered.bam nchrs比对结果,草图,预估染色体数量,三个参数就可以启动分析了,很方便;挑几个我测了的接口说一下:
- 流程默认的酶切位点是
GATC(MboI/DpnII), 如果是其他酶可以使用--RE接口调整; - 对于多倍体,或未分型的数据,我实际测试发现直接运行可能报错,加上
--remove_allelic_links参数可以解决; --correct_nrounds支持多轮自动纠错 contig, 可能会切割现有草图 (contig); 这里要注意,切割后的 fa 未必能接入你的现有流程;如果确实需要切割处理,不如自己在 juice box 里面切,结果接入下游分析更方便;
结果说明
不考虑处理中间结果的情况下,结果输出在 04.build 目录下,包括以下内容:
scaffolds.agp和scaffolds.raw.agp: 九列 agp, 主要是对于切割后的 contig 表现形式不同,这个看自己的后续分析需求吧;scaffolds.fa: 聚类完成的基因组,如果热图效果好基本可以直接用了,contig 之间有 100 bp 的 N (可调);juicebox.sh: juicer_tools 的脚本,用于生成.hic文件和.assembly文件,输入 juicer box 做后续分析或手动调整;
Quick view mode
以下几种情况可以使用 --quick_view 处理:
- 不确定样本有多少染色体
- 默认参数跑不动聚类异常(多倍体的情况)
- 仅需要跑出
.hic文件,后续希望自己调图处理
总结
感觉像是基于 ALLHiC 自己改了的软件,本身在自动化水平以及流程设计上很不错了,够"傻瓜" (褒义); 也有很多的接口供用户深度使用。
碍于我手上都是未公开数据,不能具体展示效果,只能口头描述:对于聚类软件,简单基因组各有各的优势,复杂基因组各有各的问题;对于简单二倍体植物基因组,HapHiC 相比我使用的 yahs 没有明显优势;对于多倍体基因组或包含冗余的复杂基因组,软件的效果又没有达到我的预期;
当然这也可能是我对软件使用的比较浅,比如软件提供的基于 gfa 或 p_utg.gfa 的处理模式,限于手头数据和时间关系没有进一步测试;预印文章中提到他们做了同源四倍体马铃薯的测试,要是有时间我也想把这个数据搞下来测测看。