All in one 的基因组统计工具, 主要统计了 base 信息, gap 信息, N50 信息, 以便快速了解基因组情况. Rust 编译, 运行速度还可以. 统计信息都比较基础, 写这个工具主要是图一个方便.
参数说明
程序主要有以下接口:
- -f, --fasta: 必须参数, 传入需要统计的基因组
- -o, --outdir: 可选参数, 输出文件路径, 默认当前路径 (./)
- --chr_prefix: 可选参数, 指定需要统计的染色体前缀, 默认不过滤. 可以选择只选择 chr 开头的统计, 这样就跳过 contig 不统计了
- --prefix: 可选参数, 输出文件前缀, 默认 statistics
结果说明
程序自动运行, 完成后输出四个统计文件:
- LC_Basic. txt: 基因组整体统计概览, 如下图
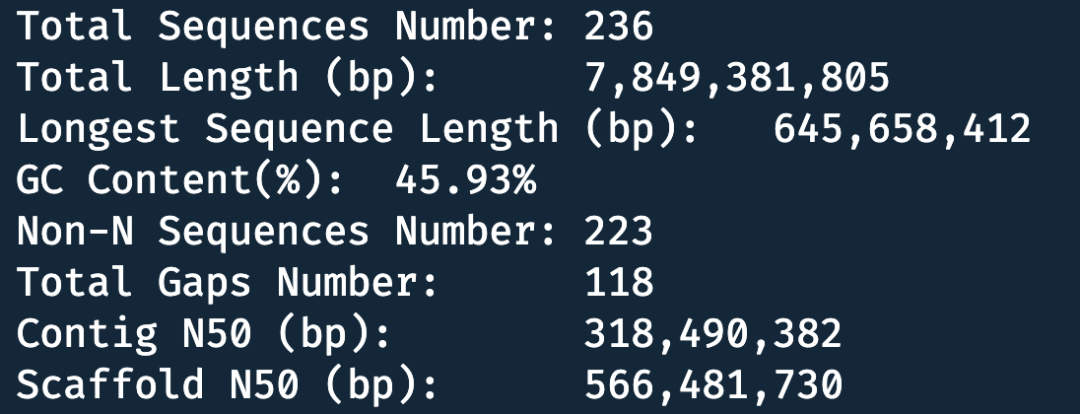
- statistics_BASE. txt: 碱基统计结果, 包含单条序列总长, A, T, G, C, N, G+C 碱基数以及碱基比例, 最后一行包含全基因组统计;
- statistics_GAP. txt: GAP统计结果, 序列中的 N 视为 gap, 统计了 gap 位置以及长度, 并生成固定格式的 id 以便使用.
- statistics_N 50. txt: N 50-N 90 统计结果, 分为 scaffold 和 contig 两类
特别说明:
- statistics_BASE. txt 中最后一行是全基因组水平的统计结果
- GAP 统计结果生成的 bed 文件是 0-base 的左闭右开格式, 即第一个碱基位置索引是 0, 以适配 bedtools
- 单条序列视为 scaffold, 若其中包含 N 碱基, 则以 N 碱基为断点生成的序列视为 contig
- 若序列 ID 包含空格或 tab 分隔, 自动选取第一个字符串作为识别 ID
- 输出结果带千分位符, 若想直接用于后续分析需要自行
sed一下
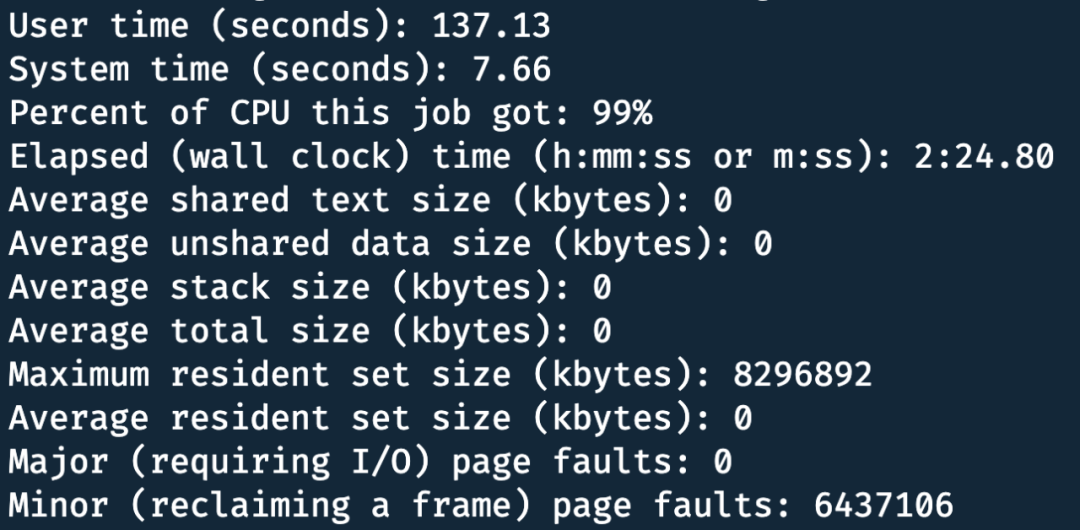
资源消耗
使用程序统计 8 G 禾草(Leymus chinensis)基因组, 资源消耗及运行时间如下:
程序下载
源码及编译好的程序发布在 GitHub 上, 欢迎下载使用.
地址:
https://github.com/WangZhSi/Bioinformatics_tool/tree/main/fasta_stat