写在前面
- 分析基于 OrthoFinder 结果, 这个工具网上教程比较多了, 这里直接使用结果文件;
- 部分代码参考发表的葡萄泛基因组文章;
- 本质是一个 R 脚本, 可视化部分使用
ggplot2, 方便大家自行修改;
接口说明
本工具包含以下接口:
-i: OrthoFinder 结果中Orthogroups/Orthogroups.GeneCount.tsv文件;-o: 输出文件目录, 文件名已经在脚本中指定, 如果目录不存在不会创建;-s: 统计 pan/core 基因集时模拟不放回抽取的次数, 默认 100;--core: 核心基因集系数, 默认为 1;--softcore: 软核心基因集系数, 默认 0.9;--private: 私有基因集系数, 默认 0.01, 最小为 1 个样本;
特别说明一下后面三个参数, 脚本自动统计输入的样本数/基因集数, 在这个基础上, 乘以相应的系数用于基因集判断, 对于默认参数, 则:
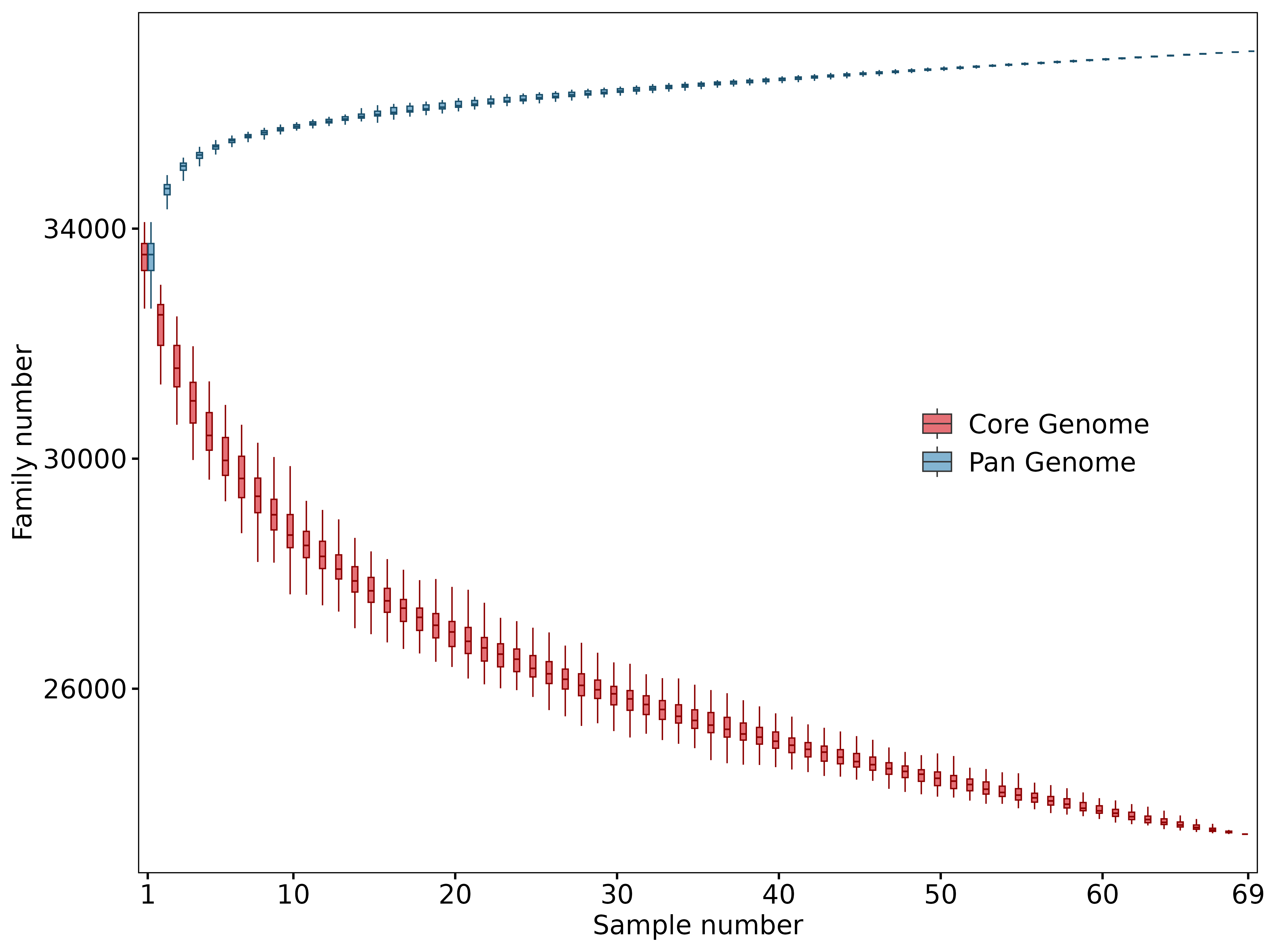
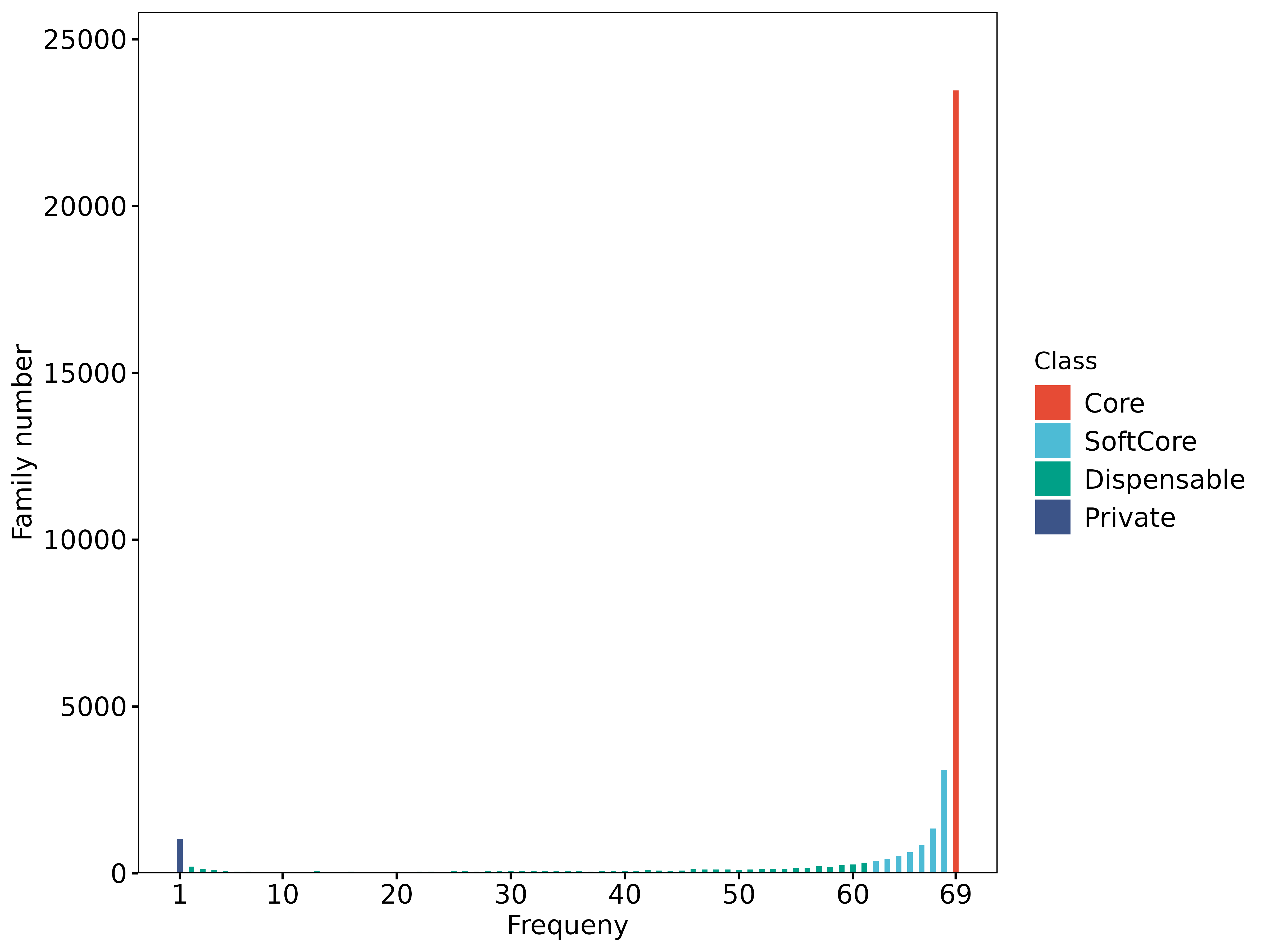
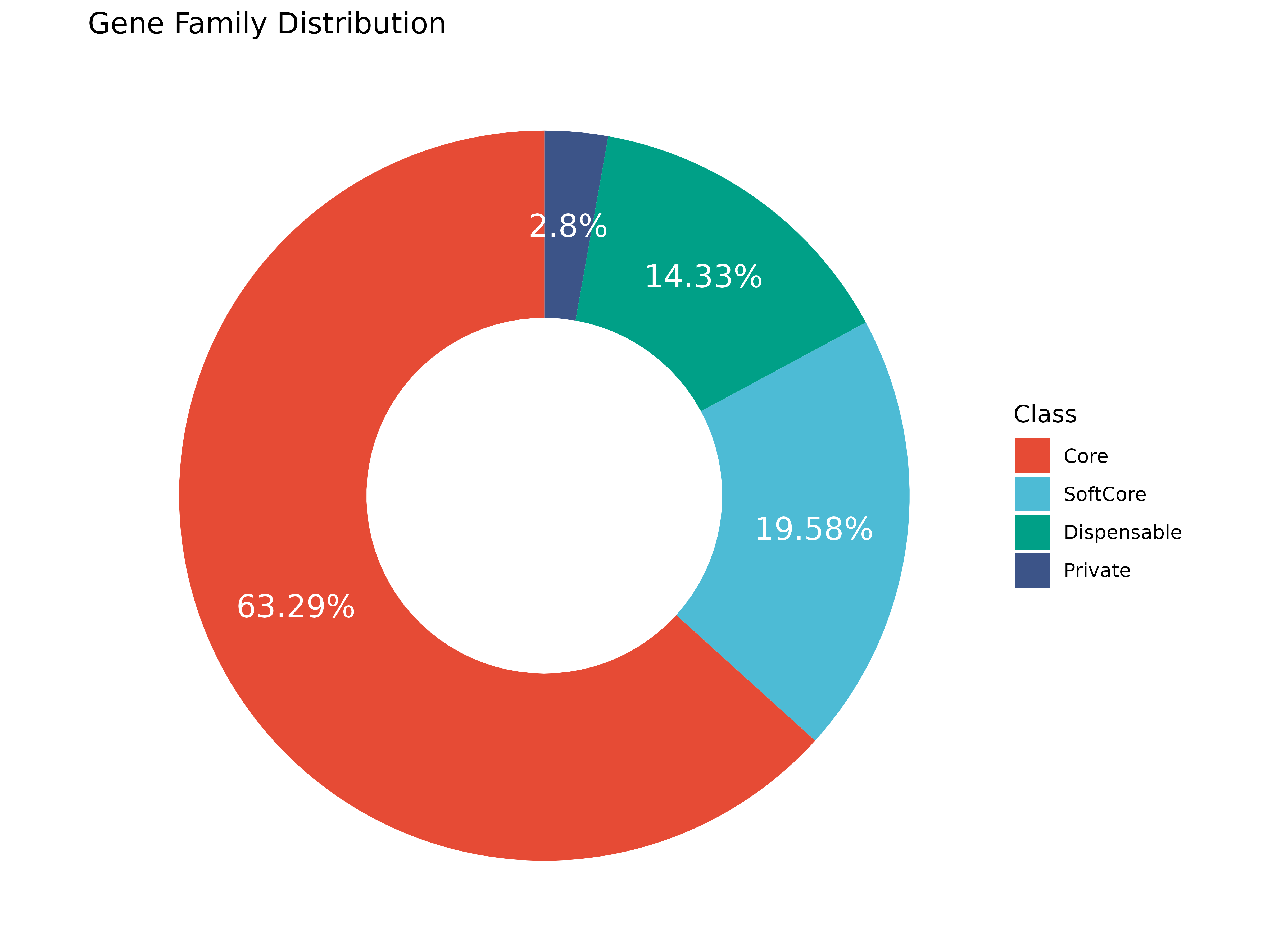
- 出现在全部样本的基因家族视为 core;
- 出现在至少 90% 样本的基因家族视为 softcore;
- 仅有 1%样本(或仅有一个样本) 支持的基因家族视为 private;
- 其余基因家族视为 dispensable;
结果说明
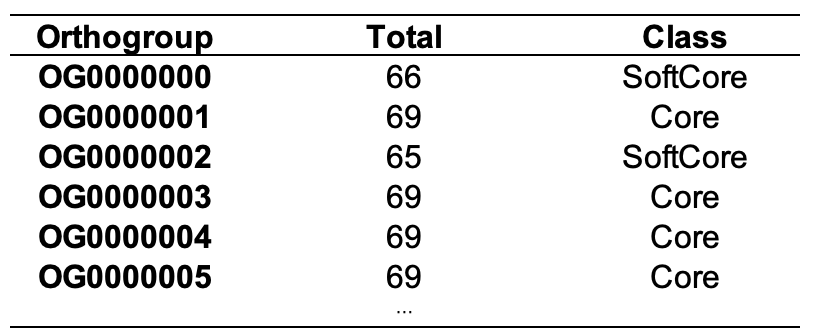
主要结果包括三个图两个表:
主要统计表:

脚本使用
脚本已经发布在 GitHub 上, 欢迎大家使用.